
Dr. Raul Duran
Research officer (CR1), Inserm
Cette adresse email est protÃĐgÃĐe contre les robots des spammeurs, vous devez activer Javascript pour la voir.
Website
Tel: +33(0) 5 40 00 22 59
Bio
Raul V. Duran obtained his PhD at the University of Seville (Spain) in 2005 on cellular bioenergetics under the supervision of Prof. Miguel Angel de la Rosa. Then, he joined the lab of Prof. Eyal Gottlieb at Cancer Research UK (Glasgow, UK) as a postdoctoral researcher (2006-2009). During this period, he worked on the metabolism of cancer cells, with especial emphasis on the role of prolyl hydroxylases as regulators of cell metabolism. Later, he moved to the lab of Prof. Michael N. Hall at the Biozentrum (University of Basel, Switzerland) as a senior postdoc (2010-2013) to study the role of the metabolism of glutamine in the control of cell growth. In June 2013, he joined the IECB as a Junior Group Leader, focusing on the crosstalk between metabolism and cell signaling in cancer cells. In 2015 he obtained a CR1 position at INSERM.
Summary
We study the mechanisms by which the de-regulation of cell signaling contributes to metabolic transformation in cancer. Particularly, we focus our research on the regulation of glutamine addiction by the mTORC1 pathway in cancer cells. First, we investigate the biochemical and cellular mechanisms of the interaction between glutaminolysis and mTORC1. Second, we study the interplay between glutamine metabolism and mTORC1 in cancer models of lymphoblastic leukemia driven by Notch upregulation. Third, we analyze the role of glutamine-dependent activation of mTORC1 in cell death and senescence in cancer models. Those investigations will permit, in collaboration with the Institut BergoniÃĐ, the implementation of clinical trials evaluating glutamine and mTOR as potential therapeutic co-targets against cancer.
Activity report
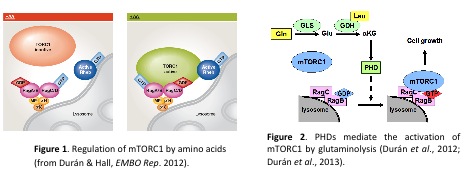
mTOR is a serine/threonine kinase highly conserved from yeast to human, which integrates several stimuli to regulate cell growth, metabolism and aging. mTOR forms two functionally and structurally distinct complexes termed mTORC1 (comprising mTOR, Raptor and mLST8) and mTORC2 (comprising mTOR, Rictor, mSIN1, PRR5 and mLST8) (DurÃĄn & Hall, 2012). Four main inputs regulate mTORC1: nutrients, growth factors, cellular bioenergetics (controlled by the AMPK pathway), and oxygen availability. Thus, mTORC1 regulates protein synthesis, ribosome biogenesis, nutrient uptake and autophagy in response to growth factors, amino acids, and cellular energy. The Rag GTPases, members of the Ras GTPase superfamily, activate mTORC1 in response to amino acids, especially leucine. The activation of Rag GTPases by amino acids is mediated by a GEF complex termed Ragulator, any by a double GAP component, termed GATOR1 and GATOR2. The addition of amino acids allows the Rag heterodimer to bind and thereby recruit mTORC1 to the lysosome, where mTORC1 is activated (Figure 1). Therefore, according to the current model, the activation of the catalytic GEF activity of Ragulator and the inhibition of the GATOR system are two key steps in the activation of mTORC1 by amino acids. To date, the molecular mechanism leading to the activation of Ragulator and GATOR by amino acids is not well understood. Of note, glutaminolysis, the deamination of glutamine to produce Îą-ketoglutarate (ÎąKG), is a critical step in the Rag-dependent recruitment of mTORC1 to the lysosome (DurÃĄn et al., 2013), but the molecular mechanism connecting glutaminolysis and mTORC1 has not been clarified yet.
Glutamine, plays a particularly important role in cell growth and metabolism as a precursor for ÎąKG of the tricarboxylic acid cycle, for nucleotides and for other amino acids. Glutamine is metabolized through glutaminolysis, which consists of two steps. The first step, catalyzed by the enzyme glutaminase (GLS), converts glutamine to glutamate. The second step, catalyzed by glutamate dehydrogenase (GDH), converts glutamate to ÎąKG. In the past, we demonstrated that glutaminolysis activates mTORC1 by enhancing glutaminolysis and ÎąKG production (DurÃĄn et al., 2012; DurÃĄn & Hall, 2012). We also described that the prolyl hydroxylases domain (PHD) proteins are necessary to induce mTORC1 activation by glutaminolysis (Duran et al., 2013). Thus, ÎąKG produced by glutaminolysis activates PHD, which in turn induce the lysosomal translocation of mTORC1 and its subsequent activation (Figure 2).
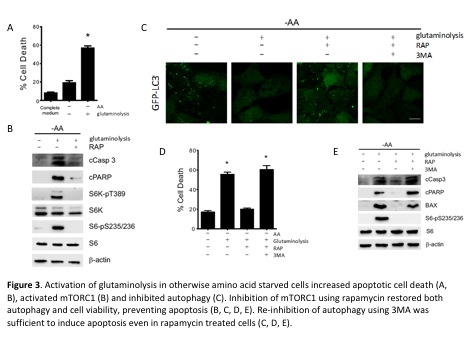
Unbalanced activation of glutaminolysis and cell death
Despite the role of both glutaminolysis and mTORC1 in the promotion of cell growth, we recently observed that the unbalanced activation of the glutaminolysis/mTORC1 pathway in the absence of other amino acids induced apoptotic cell death in human cells. Indeed, the long-term production of glutaminolysis-derived ÎąKG in the absence of other amino acids was sufficient to activate mTORC1 and to inhibit autophagy, but concomitantly reduced cell viability and activated apoptosis. Inhibition of mTORC1 or re-activation of autophagy were sufficient to suppress the glutaminolysis-induced apoptosis (Figure 3). We additionally observed that the ability of rapamycin to prevent apoptosis resides in its pro-autophagic potential (Villar et al., 2015): re-inhibition of autophagy prevented cell survival in rapamycin-treated cells. In agreement with our results, previous reports have shown that increasing intracellular ÎąKG levels induces apoptosis, although no clear mechanism was provided (Tennant et al., 2009; Tennant and Gottlieb, 2010). In the other hand, a recent report has shown that glutaminolysis is crucial for the induction of ferroptosis, a non-apoptotic type of cell death (Gao et al., 2015). The surprising role of glutaminolysis as a cell death inducing mechanism during nutrient restriction points at the importance of nutritional imbalance in the control of cell viability and the potential use of this metabolic disequilibrium to identify new metabolic components and targets with potential implication in the treatment of nutrition-related diseases, such as obesity, diabetes, cancer, or cardiovascular diseases. In any case, whether the activation of apoptosis by the unbalanced activation of glutaminolysis and mTORC1 plays an active role in the biochemical basis of those diseases is a question that remains to be elucidated.
Selected Publications
- TerÃĐs S, Nguyen TL and DurÃĄn RV (2016) Metabolic transformation in Notch-driven acute lymphoblastic leukemia, Int. J. Mol. Biol. Med., in press.
- Klionsky DJ et al. (2016) Guidelines for the use and interpretation of assays for monitoring autophagy, Autophagy 12, 1-222.
- Villar VH, Mehri F, Djavaheri-Mergny M and DurÃĄn RV (2015) Glutaminolysis and autophagy in cancer, Autophagy 11, 1198-208.
- DurÃĄn RV, MacKenzie EM, Boulahbel H, Frezza C, Tardito S, Heiserich L, Bussolati O, Rocha S, Hall MN and Gottlieb E (2013) HIF-independent role of prolyl hydroxylases in the cellular response to amino acids. Oncogene 32, 4549-56.
- DurÃĄn RV, Oppliger W, Robitaille AM, Heiserich L, Skendaj R, Gottlieb E and Hall MN (2012) Glutaminolysis activates Rag-mTORC1 signaling. Mol. Cell 47, 349-58.
Research Team
- Dr. Raul DURAN, Team Leader
- Dr. Victor VILLAR, postdoc (CRA - SIRIC/BRIO)
- Tra Ly NGUYEN, PhD student (CRA)
- ClÃĐment BODINEAU, M2 Student (Univ. Bordeaux)
- Sarah COURTOIS, Visitor PhD (Univ. Bordeaux)
- Marion MULLER, Undergraduated Student (Univ. Bordeaux)
This team is part of the unit U1218 ACTION, Inserm/ Institut BergoniÃĐ
|



